بیماری مننژیوما
خلاصه مقاله
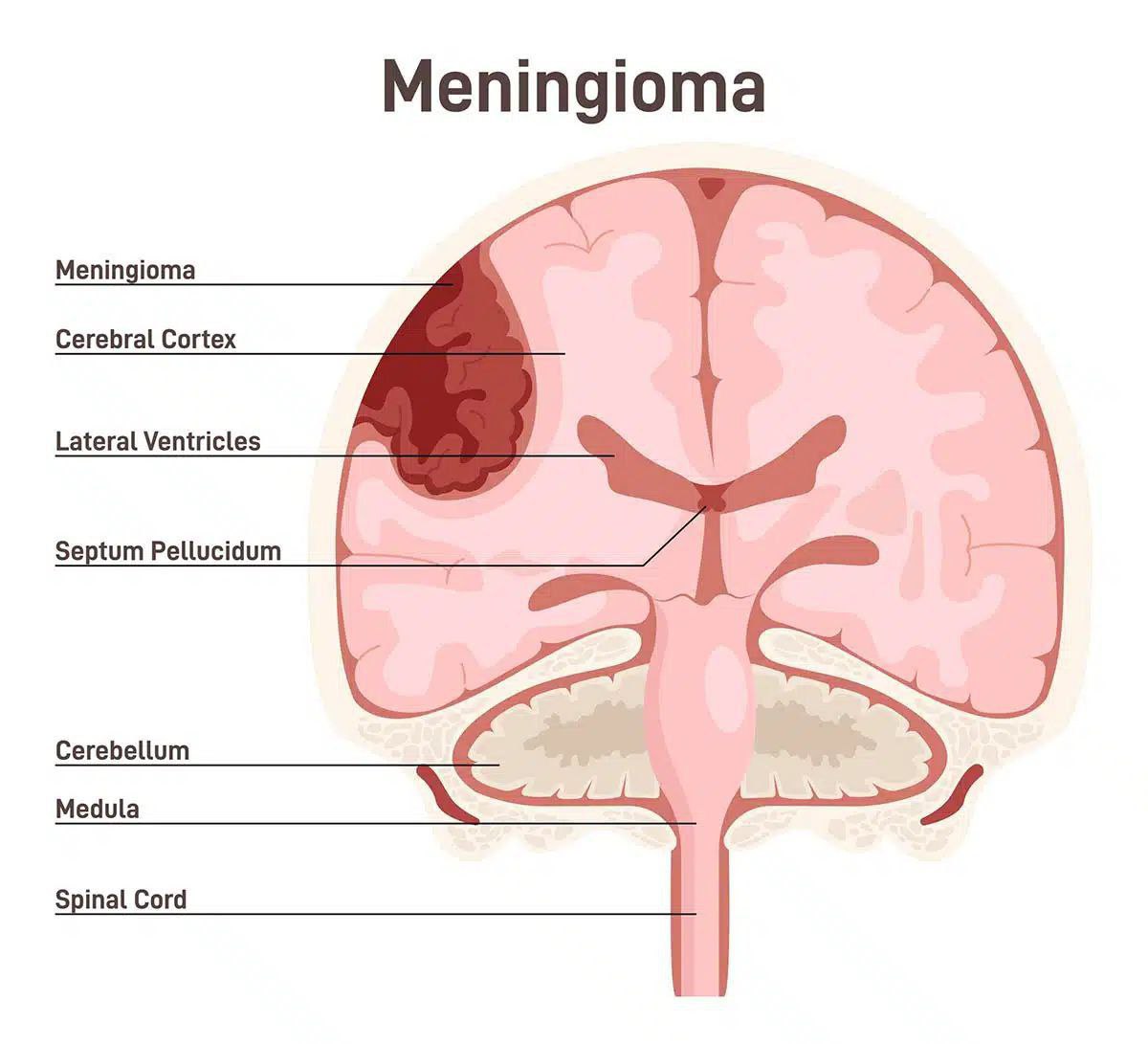
مننژیوما یک نوع تومور مغزی است که از بافت های مننژهای مغز (پوششهای مغز) تشکیل شده است. این تومورها معمولاً از بافتهای پوششی مغز (مننژها) شروع میشوند و میتوانند به شکلهای مختلفی در مغز ظاهر گردند.
#با_مقاله_درس_بخوانیم
#مننژیوما
🔰مننژیوما یک نوع تومور مغزی است که از بافت های مننژهای مغز (پوششهای مغز) تشکیل شده است. این تومورها معمولاً از بافتهای پوششی مغز (مننژها) شروع میشوند و میتوانند به شکلهای مختلفی در مغز ظاهر گردند.
🔰مننژیوماها میتوانند در سه نوع اصلی ظاهر شوند: مننژیومای خوش خیم (بیشترین تعداد)، مننژیومای خوش خیم با تغییرات بدخیم و مننژیومای بدخیم (کمترین تعداد):
1. مننژیومای خوش خیم: این نوع تومور مننژیوما بیشترین تعداد را تشکیل میدهد. آنها معمولاً از بافتهای پوششی مغز (مننژها) شروع میشوند و به طور عمومی رشد کنندگی آهستهتری دارند. این نوع تومورها در بیشتر موارد خوش خیم هستند و اغلب قابل درمان می باشند.
2. مننژیومای خوش خیم با تغییرات بد خیم: این نوع تومورها نیز از بافتهای پوششی مغز (مننژها) شروع میشوند، اما دارای تغییرات بد خیم بیشتری هستند. این تغییرات ممکن است شامل رشد سریعتر تومور، نفوذ به بافتهای اطراف مغز و احتمال بالاتر عود تومور پس از درمان باشد.
3. مننژیومای بدخیم: این نوع تومورها کمترین تعداد را تشکیل میدهند و بیشترین خطر را دارند. آنها معمولاً سریعتر رشد میکنند و به بافتهای اطراف مغز نفوذ میکنند. درمان مننژیومای بدخیم ممکن است شامل جراحی، رادیوتراپی و شیمی درمانی باشد.
🔰علائم و نشانههای مننژیوما ممکن است شامل سردرد، تاری دید، تشنج، کاهش قدرت حافظه، تغییرات در رفتار و اختلالات حرکتی باشد. لازم است بدانید که تشخیص و درمان مننژیوماها بستگی به مشخصات هر بیمار و ویژگیهای تومور دارد.
🔰برای تشخیص مننژیوما، انجام تصویربرداری مغزی مانند MRI و CT scan ضروری است. درمان مننژیوما معمولاً شامل جراحی برای برداشتن تومور، رادیوتراپی و/یا شیمی درمانی است.
✍پدیدآورنده: دکتر مریم سرحدی
📚منبع:
-Principles and Practice of Neuro-Oncology
-Handbook of Neuro-Oncology Neuroimaging
✨شکوه دنیای اعصاب در انجمن علمی دانشجویی جراحی اعصاب
با ما همراه باشید.✨
🆔English:@NeurosurgeryAssociation
🆔Persian:@Neurosurgery_Association
مقالات مرتبط
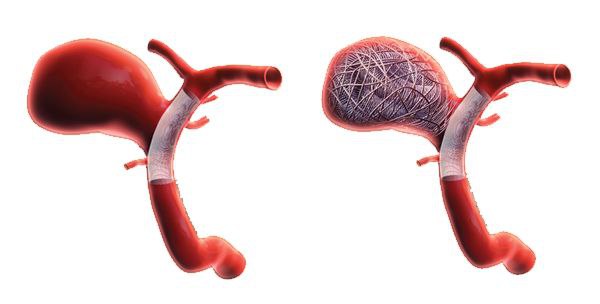
ابزار های جدید راه های درمان آنوریسم را گسترش میدهند
تصمیم درباره درمان آنورسیم های داخل مغز بسیار دشوار میباشد که نیازمند این است که عوارض و فواید عمل برای هر بیمار بادقت بررسی شود.
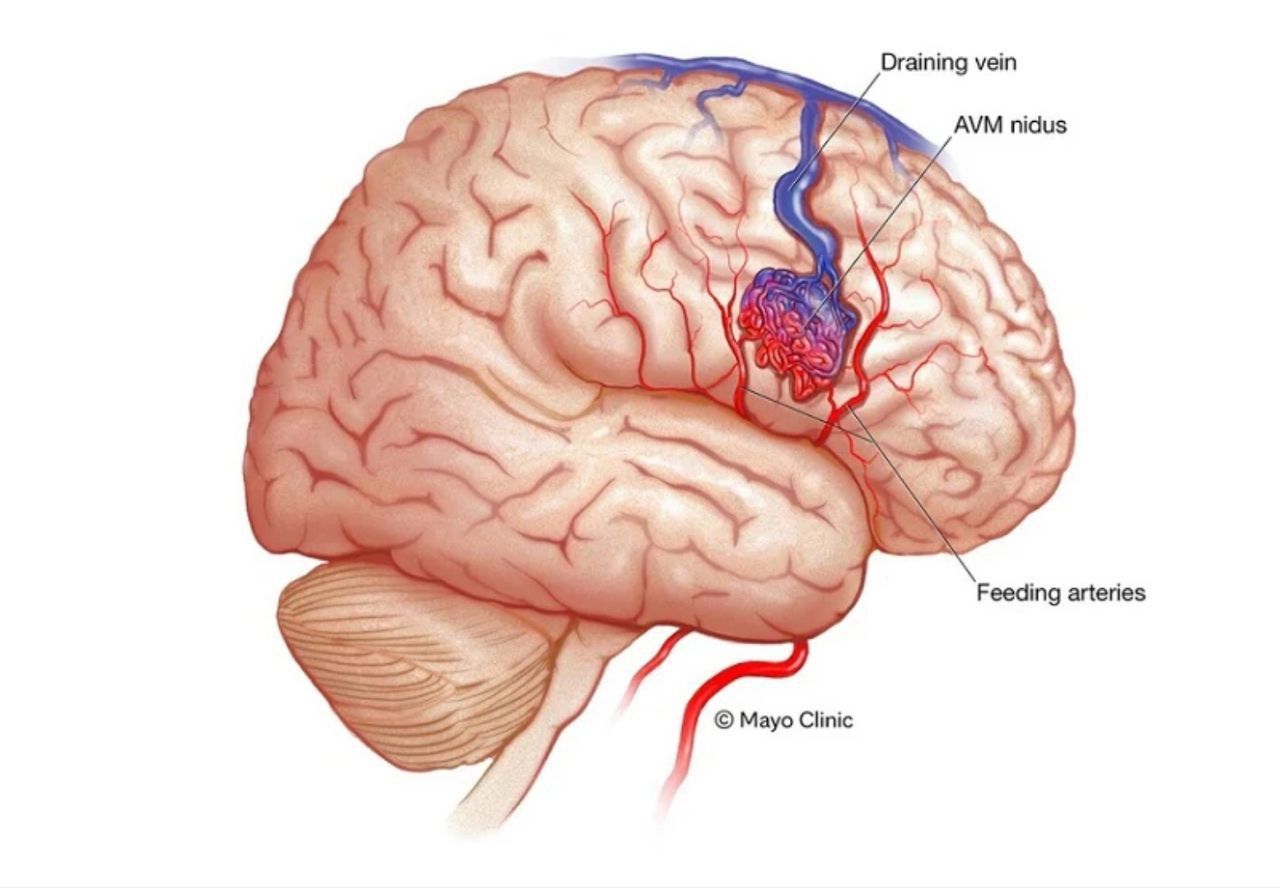
ملفورماسیون_شریانی_وریدی
مالفورمیشن های شریانی وریدی یا به اختصار AVM بسیار متفاوتی وجود دارند که راه های درمان دگرگونی نیز برای آنها یافت میشود